医疗器械产品注册技术审评报告
产品中文名称:取栓支架
产品管理类别:第三类
申请人名称:珠海通桥医疗科技有限公司
国家药品监督管理局
医疗器械技术审评中心
基本信息
一、申请人名称:珠海通桥医疗科技有限公司
二、申请人住所:珠海市香洲区唐家湾镇科技七路1号珠海中电高科技产业园4栋1-B单元
三、生产地址:珠海市国家高新技术开发区科技七路中电高科技园四栋1B
产品审评摘要
一、产品概述
(一)产品结构及组成
该产品由一个自扩张的取栓支架、输送丝、支撑弹簧圈、显影弹簧圈、热缩套管、显影环和保护鞘管组成。其中取栓支架和输送丝均选用镍钛合金材料制成,显影环材质为铂铱合金,显影弹簧圈材质为铂钨合金。产品采用环氧乙烷灭菌,一次性使用。货架寿命三年。
(二)产品适用范围
用于在患者缺血性卒中发作8小时内移除堵塞在颅内大动脉血管内的血栓以达到恢复血流的治疗目的,包括颈内动脉、大脑中动脉的M1和M2段、大脑前动脉的A1和A2段。
(三)型号/规格
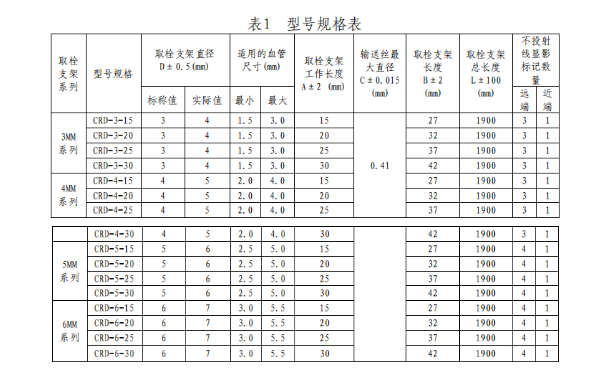
(四)工作原理
取栓支架通过微导管技术,沿体内动脉通道到达颅内血栓栓塞位置后,取栓支架推出微导管自膨胀张开,建立即刻的血流通路,使处于危险边缘的脑细胞得到再灌注,同时,回撤取栓支架和微导管系统,将被其捕获到支架网眼内的血栓移除出体外,恢复血流通畅。
二、临床前研究摘要
(一)产品性能研究
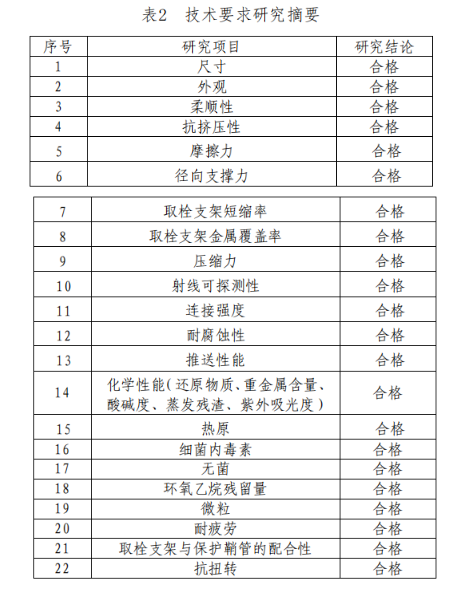
1.产品技术要求
研究技术要求研究项目如表2所示:
2.产品性能评价
产品性能评价包括模拟取栓、取栓支架在微导管内摩擦力、贴壁性能、不同压缩状态下径向力比较以及产品技术要求项目等研究,结果表明产品符合设计输入要求。
(二)生物相容性
该产品与人体接触方式为短期外部接入,接触部位为循环血液,接触时间为24小时内,申请人依据GB/T 16886系列标准进行了生物相容性评价,申请人提供了细胞毒性、迟发型超敏反应、皮内刺激、急性全身毒性、溶血试验、部分凝血激活酶时间试验、体内血栓形成检验报告,产品生物相容性风险可接受。
(三)灭菌
该产品采用环氧乙烷灭菌,无菌状态提供。申请人提供了灭菌过程确认报告,证明无菌保证水平可达10-6。环氧乙烷残留量不大于10μg/g,2-氯乙醇残留水平不超过9mg/件。
(四)产品有效期和包装
该产品货架有效期三年。申请人提供了货架有效期验证报告,验证实验为加速老化和实时老化验证,包括产品稳定性、包装完整性验证。提供了产品运输稳定性验证资料。
(五)动物研究
该研究的目的是评价取栓支架在急性缺血性动物模型体内采用机械取栓的方法取出栓塞的血栓以达到恢复血流为治疗目的的安全性和有效性。
实验在健康比格犬身上建立血栓栓塞动物模型,在取栓即时、30天、90天评价器械操作性(取栓支架的推送性能、柔顺性、可靠性和X-射线下的显影性,包括限定取栓支架的安全取栓次数和取栓时间)、安全性(取栓过程中的不良事件、潜在的血管损伤、出血、血栓形成、取栓过程对血管壁的损伤等)、有效性(取栓效果、取栓前后血流恢复效果、取栓过程中小血栓形成或远端血管闭塞的情况)、血管造影分析等。实验结果表明产品可达到预期设计要求。
三、临床评价摘要
该产品以临床试验方式进行临床评价。临床试验目的为为评价申报产品用于患者大动脉闭塞导致的急性缺血性脑卒中发作8小时内移除堵塞在颅内血管内颈内动脉(ICA)、大脑前动脉A1段和A2段、和大脑中动脉M1和M2段的血栓的安全性和有效性。临床试验采用前瞻性、多中心、随机对照、非劣效检验设计,对照组为血流重建装置SolitaireFR(国械注进20173776118)。
该临床试验在17家临床机构开展,实际入组217例(试验组107例/对照组110例)患者。主要研究终点为术中血管成功再通率,次要指标包括术中成功再通的血管比率、血管再通时间、术后24小时和术后7天NIHSS评分、术后90天mRS评分及0-2分的比率、输送性能评价,安全性评价指标包括24小时症状性颅内出血发生率、24小时蛛网膜下腔出血发生率、严重不良事件发生率、不良事件发生率、器械缺陷发生率、24小时内非症状性颅内出血率、24小时内死亡率、脑疝、实质性出血I型、实质性出血II型、症状性和非症状性脑出血。
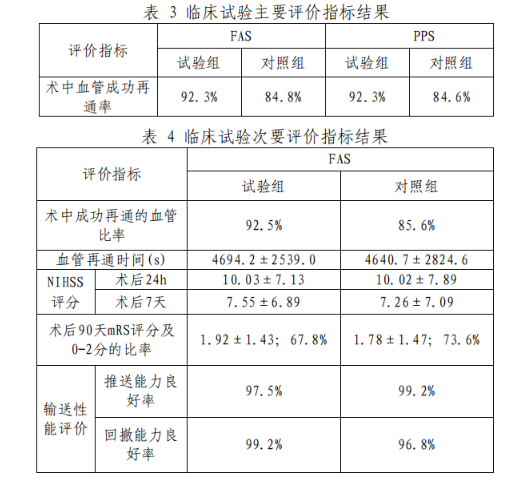
全分析集(FAS)包含患者209例,其中试验组104例,对照组105例;符合方案集(PPS)包含患者208例,其中试验组104例,对照组104例;安全集(SS)包含患者217例,其中试验组107例,对照组110例。主要有效性指标结果如表3所示,次要有效性指标结果如表4所示。结果表明两组差值的95%可信区间下限大于-12%,95%可信区间估计表明非劣性研究假设成立。
![]()
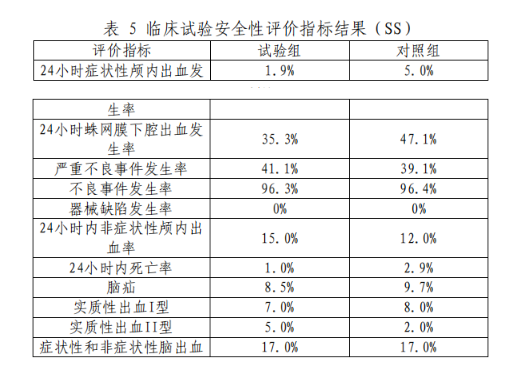
安全性指标分析如表5所示,24小时症状性颅内出血发生率、24小时蛛网膜下腔出血发生率、严重不良事件发生率、不良事件发生率、器械缺陷发生率、24小时内非症状性颅内出血率、24小时内死亡率、脑疝、实质性出血I型、实质性出血II型、症状性和非症状性脑出血无统计学差异。
![]()
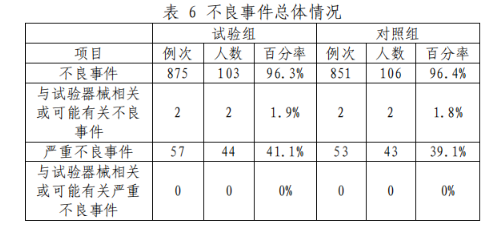
不良事件发生情况如表6所示,试验组与对照组间无统计学差异,与器械相关性分析结果为试验组与对照组间无统计学差异。
该产品在临床应用中通过移除患者大血管病变的血栓实现血管再通,临床上对卒中发生8小时内的患者进行治疗,挽救患者生命或降低患者身体伤害的风险。但可能有潜在的并发症,如穿刺部位血肿和出血、血管壁穿孔或剥离、血管痉挛、包括中风和死亡的神经功能恶化、局部缺血、远端血管再次闭塞、器械变形、折叠、断裂或发生故障、血栓症(急性和亚急性)、缺血或颅内出血、假性动脉瘤形成和动静脉瘘。根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品为患者带来的临床受益大于风险。
四、风险分析及说明书提示
参照《YY/T0316-2016医疗器械风险管理对医疗器械的应用》,对该产品进行风险分析。对目前已知及可预测风险采取了风险控制措施,经综合评价,在目前认知水平上,认为该产品上市带来的获益/受益大于风险。为保证用械安全,需在说明书中提示以下信息:
(一)明确的产品适用范围
(二)警告及预防措施
1.取栓支架仅供受过介入性神经放射学和缺血性卒中介入治疗训练的医生使用。
2.取栓支架的某些部件是很纤弱的,需要仔细地操作,任何不当的操作手法都有损坏器械的可能。
3.切勿使用过大的力推进输送丝。当未确定阻力的原因时,切勿盲目推进取栓支架,应先稍稍回撤后再往前推进。如未能查明原因或尝试数次失败后,应退出取栓支架检查是否有损坏。
4.切勿大力扭转取栓支架。
5.为减少血管损伤,在同一血管内,一般不超过3次取栓操作,且不同血管内严禁使用同一装置进行2次以上的取栓操作。
6.当取栓支架自膨胀完全展开后,切勿在人体血管内调整取栓支
架的位置或回撤进微导管,应推送微导管直至抵住取栓支架,然后将取栓支架与微导管作为一个整体一起回撤至导引导管或撤出体外。取栓支架必须回收到微导管内,才可在所需位置重新展开或从患者体内取出。
7.每一根取栓支架必须配用一根新的微导管。
8.取栓支架以无菌包装方式提供,仅限一次性使用,严禁二次灭菌后再次使用。
9.使用完后,废弃物须根据医院、管理部门或当地政府的政策进行处理。
10.储藏:本产品必须储藏在干燥、清洁、通风良好、无腐蚀性气体的环境中。
11.在标识的有效期内使用本产品。
12.切勿使用包装已破损的产品。
13.切勿将本产品采用高温蒸汽灭菌。
14.切勿将本产品暴露在有机溶剂中,这将导致某些连接损坏。
15.一个小的圆灭菌指示标签贴在产品包装上,以便在无菌屏障被破坏前可以看见。此指示标签暴露在环氧乙烷下将由红变蓝。因此,如果该灭菌指示标签为红色,则不要使用该器械。
16.使用前,应先用0.9%的生理盐水冲洗本器械,并使其一直保持湿润。
(三)禁忌症
取栓支架可能的禁忌症包括但不限于以下几种:
1.已知对镍钛有过敏反应者。
2.禁用抗凝血药、抗血小板治疗法或血栓溶解药的患者
3.患者的造影显示有颈动脉夹层
4.患者入路显示有粥样硬化导致的闭塞,导致取栓支架无法安全地通过与回收。
5.不能配合和耐受介入手术者。
综合评价意见
该申报产品属按照《创新医疗器械特别审批程序(试行)》审批项目,编号201700074。申请人的注册申报资料符合现行要求。依据《医疗器械监督管理条例》(国务院令第680号)、《医疗器械注册管理办法》(原国家食品药品监督管理总局令2014年第4号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。
2020年9月1日
![]()
© 2018 - 2020, Wuhan Tacro Technology Co.,Ltd All Rights Reserved.

